The neural basis of sensory hypersensitivity
A new study may explain why people with autism are often highly sensitive to light and noise.

Many people with autism spectrum disorders are highly sensitive to light, noise, and other sensory input. A new study in mice reveals a neural circuit that appears to underlie this hypersensitivity, offering a possible strategy for developing new treatments.
MIT and Brown University neuroscientists found that mice lacking a protein called Shank3, which has been previously linked with autism, were more sensitive to a touch on their whiskers than genetically normal mice. These Shank3-deficient mice also had overactive excitatory neurons in a region of the brain called the somatosensory cortex, which the researchers believe accounts for their over-reactivity.
There are currently no treatments for sensory hypersensitivity, but the researchers believe that uncovering the cellular basis of this sensitivity may help scientists to develop potential treatments.
“We hope our studies can point us to the right direction for the next generation of treatment development,” says Guoping Feng, the James W. and Patricia Poitras Professor of Neuroscience at MIT and a member of MIT’s McGovern Institute for Brain Research.
Feng and Christopher Moore, a professor of neuroscience at Brown University, are the senior authors of the paper, which appears today in Nature Neuroscience. McGovern Institute research scientist Qian Chen and Brown postdoc Christopher Deister are the lead authors of the study.
Too much excitation
The Shank3 protein is important for the function of synapses — connections that allow neurons to communicate with each other. Feng has previously shown that mice lacking the Shank3 gene display many traits associated with autism, including avoidance of social interaction, and compulsive, repetitive behavior.
In the new study, Feng and his colleagues set out to study whether these mice also show sensory hypersensitivity. For mice, one of the most important sources of sensory input is the whiskers, which help them to navigate and to maintain their balance, among other functions.
The researchers developed a way to measure the mice’s sensitivity to slight deflections of their whiskers, and then trained the mutant Shank3 mice and normal (“wild-type”) mice to display behaviors that signaled when they felt a touch to their whiskers. They found that mice that were missing Shank3 accurately reported very slight deflections that were not noticed by the normal mice.
“They are very sensitive to weak sensory input, which barely can be detected by wild-type mice,” Feng says. “That is a direct indication that they have sensory over-reactivity.”
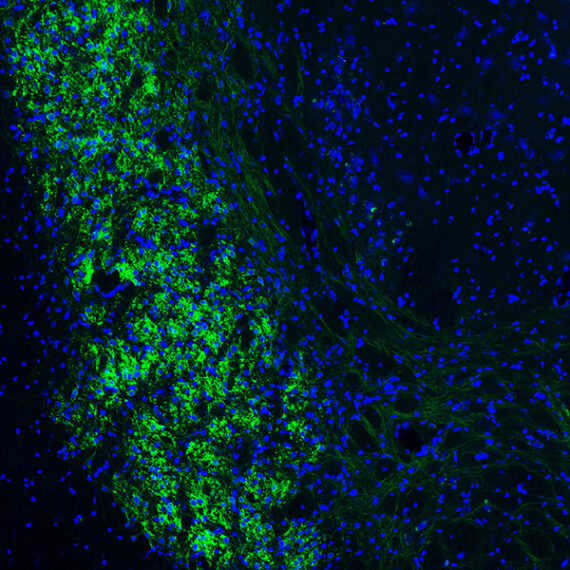
Once they had established that the mutant mice experienced sensory hypersensitivity, the researchers set out to analyze the underlying neural activity. To do that, they used an imaging technique that can measure calcium levels, which indicate neural activity, in specific cell types.
They found that when the mice’s whiskers were touched, excitatory neurons in the somatosensory cortex were overactive. This was somewhat surprising because when Shank3 is missing, synaptic activity should drop. That led the researchers to hypothesize that the root of the problem was low levels of Shank3 in the inhibitory neurons that normally turn down the activity of excitatory neurons. Under that hypothesis, diminishing those inhibitory neurons’ activity would allow excitatory neurons to go unchecked, leading to sensory hypersensitivity.
To test this idea, the researchers genetically engineered mice so that they could turn off Shank3 expression exclusively in inhibitory neurons of the somatosensory cortex. As they had suspected, they found that in these mice, excitatory neurons were overactive, even though those neurons had normal levels of Shank3.
“If you only delete Shank3 in the inhibitory neurons in the somatosensory cortex, and the rest of the brain and the body is normal, you see a similar phenomenon where you have hyperactive excitatory neurons and increased sensory sensitivity in these mice,” Feng says.
Reversing hypersensitivity
The results suggest that reestablishing normal levels of neuron activity could reverse this kind of hypersensitivity, Feng says.
“That gives us a cellular target for how in the future we could potentially modulate the inhibitory neuron activity level, which might be beneficial to correct this sensory abnormality,” he says.
Many other studies in mice have linked defects in inhibitory neurons to neurological disorders, including Fragile X syndrome and Rett syndrome, as well as autism.
“Our study is one of several that provide a direct and causative link between inhibitory defects and sensory abnormality, in this model at least,” Feng says. “It provides further evidence to support inhibitory neuron defects as one of the key mechanisms in models of autism spectrum disorders.”
He now plans to study the timing of when these impairments arise during an animal’s development, which could help to guide the development of possible treatments. There are existing drugs that can turn down excitatory neurons, but these drugs have a sedative effect if used throughout the brain, so more targeted treatments could be a better option, Feng says.
“We don’t have a clear target yet, but we have a clear cellular phenomenon to help guide us,” he says. “We are still far away from developing a treatment, but we’re happy that we have identified defects that point in which direction we should go.”
The research was funded by the Hock E. Tan and K. Lisa Yang Center for Autism Research at MIT, the Stanley Center for Psychiatric Research at the Broad Institute of MIT and Harvard, the Nancy Lurie Marks Family Foundation, the Poitras Center for Psychiatric Disorders Research at the McGovern Institute, the Varanasi Family, R. Buxton, and the National Institutes of Health.