New clues to brain changes in Huntington’s disease
Opioid receptor changes discovered in neurodegenerative disease model.
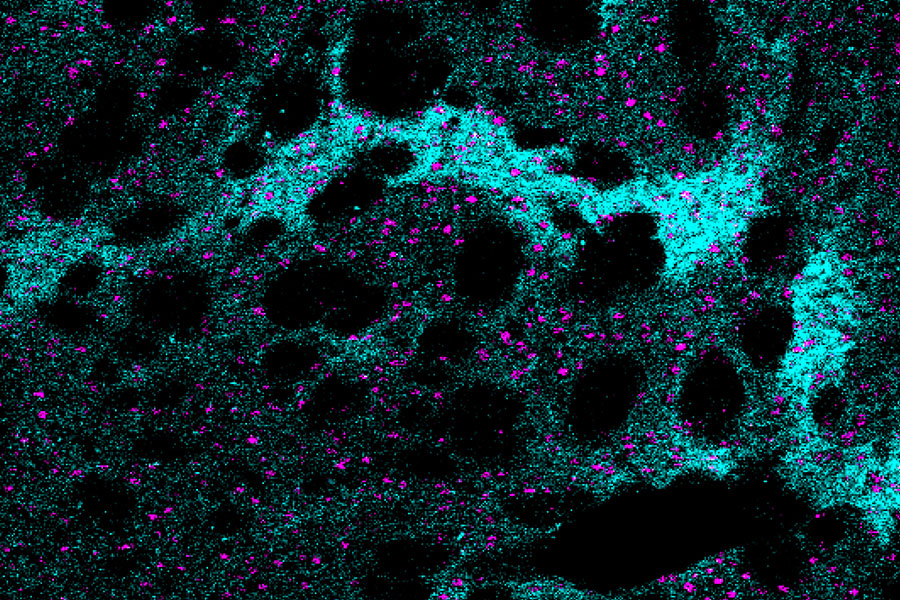
Huntington’s disease is a fatal inherited disorder that strikes most often in middle age with mood disturbances, uncontrollable limb movements, and cognitive decline. Years before symptom onset, brain imaging shows degeneration of the striatum, a brain region important for the rapid selection of behavioral actions. As the striatal neurons degenerate, their “identity” proteins, the building blocks that give particular cell types their unique function, are gradually turned off.
A new study from the lab of Institute Professor Ann Graybiel has found a surprising exception to this rule. The researchers discovered that in mouse models of Huntington’s disease, the cell identity protein MOR1, named as the Mu type Opioid Receptor, actually becomes more abundant as the striatal neurons degenerate.
“This is one of the most striking immunohistochemical change that I have ever seen in the literature of Huntington’s disease model animals,” says Ryoma Morigaki, a research scientist in the Graybiel laboratory and lead author of the report, who worked with Tomoko Yoshida and others in the Graybiel lab.
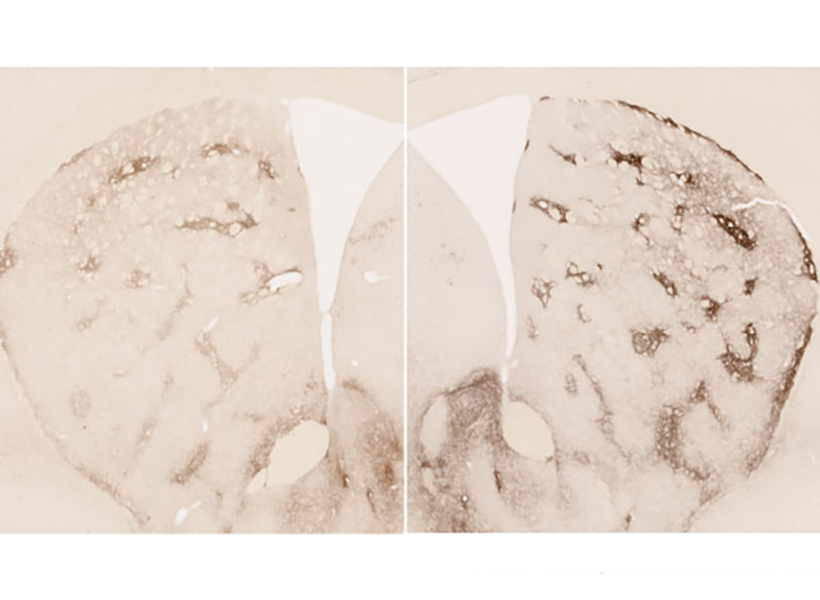
More opioid receptors
MOR1 is a receptor on the surface of neurons that binds to opioids that are produced by the body or those taken for pain relief, such as morphine. The natural opioid in the brain is a small molecule called enkephalin, and it is normally produced by the same striatal neurons that degenerate in the earliest stages of Huntington’s disease.
The research team speculates that the striatum increases the quantity of MOR1 receptors in Huntington’s disease models to compensate for plummeting levels of enkephalin, but they also believe this upregulation may play a role in the perception of reward.
Previous work suggests that MOR1 has distinct signaling mechanisms related to its function in pain perception and its function in drug-seeking. These distinct mechanisms might be related to the fact that MOR1 is produced as multiple “isoforms,” slight variations of a protein that can be read out from the same gene. The MOR1 isoform that is found in the striatum is thought to be more important for drug-seeking behaviors than for pain perception. This in turn means that MOR1 might play a role in a key striatal function, which is to learn what actions are most likely to lead to reward.
“It is now recognized that mood disturbances can pre-date the overt motor abnormalities of Huntington’s patients by many years. These can even be the most disturbing symptoms for patients and their families. The finding that this receptor for opioids becomes so elevated in mood-related sites of the striatum, at least in a mouse model of the disorder, may give a hint to the underlying circuit dysfunction leading to these problems,” says Ann Graybiel.
Clues for treatment
MOR1 is used as a standard to identify subsets of neurons that are located within small clusters of neurons in the striatum that were previously discovered by Ann Graybiel and named striosomes.
“The most exciting point for me is the involvement of striatal compartments [striosomes] in the pathogenesis of Huntington’s disease,” says Morigaki, who has now moved to the University of Fukoshima in Japan and is a practicing neurosurgeon who treats movement disorders.
MOR1-positive striosomal neurons are of high interest in part because they have direct connections to the same dopamine-producing neurons that are thought to degenerate in Parkinson’s disease. Whereas Parkinson’s disease is characterized by a loss of dopamine and loss of movement, Huntington’s disease is characterized by ups and downs in dopamine and excessive movements. In fact, the only drugs that are FDA-approved to treat Huntington’s disease are drugs that minimize dopamine release, thereby working to dampen the abnormal movements. But these treatments come with potentially severe side-effects such as depression and suicide.
This latest discovery might provide mechanistic clues to dopamine fluctuations in Huntington’s disease and provide avenues for more specific treatments.
This research was funded by the CHDI Foundation (A-5552), Broderick Fund for Phytocannabinoid Research at MIT, NIH/NIMH R01 MH060379, the Saks Kavanaugh Foundation, JSPS KAKENHI Grants #16KK0182, 17K10899 and 20K17932 , Dr. Tenley Albright, Kathleen Huber, and Dr. Stephan and Mrs. Anne Kott.