When we make complex decisions, we have to take many factors into account. Some choices have a high payoff but carry potential risks; others are lower risk but may have a lower reward associated with them.
A new study from MIT sheds light on the part of the brain that helps us make these types of decisions. The research team found a group of neurons in the brain’s striatum that encodes information about the potential outcomes of different decisions. These cells become particularly active when a behavior leads a different outcome than what was expected, which the researchers believe helps the brain adapt to changing circumstances.
“A lot of this brain activity deals with surprising outcomes, because if an outcome is expected, there’s really nothing to be learned. What we see is that there’s a strong encoding of both unexpected rewards and unexpected negative outcomes,” says Bernard Bloem, a former MIT postdoc and one of the lead authors of the new study.
Impairments in this kind of decision-making are a hallmark of many neuropsychiatric disorders, especially anxiety and depression. The new findings suggest that slight disturbances in the activity of these striatal neurons could swing the brain into making impulsive decisions or becoming paralyzed with indecision, the researchers say.
Rafiq Huda, a former MIT postdoc, is also a lead author of the paper, which appears in Nature Communications. Ann Graybiel, an MIT Institute Professor and member of MIT’s McGovern Institute for Brain Research, is the senior author of the study.
Learning from experience
The striatum, located deep within the brain, is known to play a key role in making decisions that require evaluating outcomes of a particular action. In this study, the researchers wanted to learn more about the neural basis of how the brain makes cost-benefit decisions, in which a behavior can have a mixture of positive and negative outcomes.
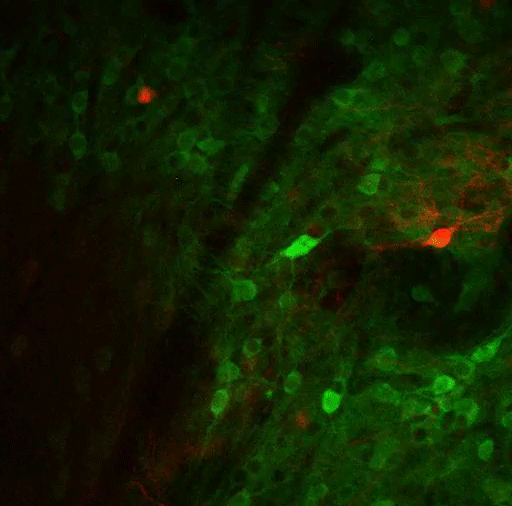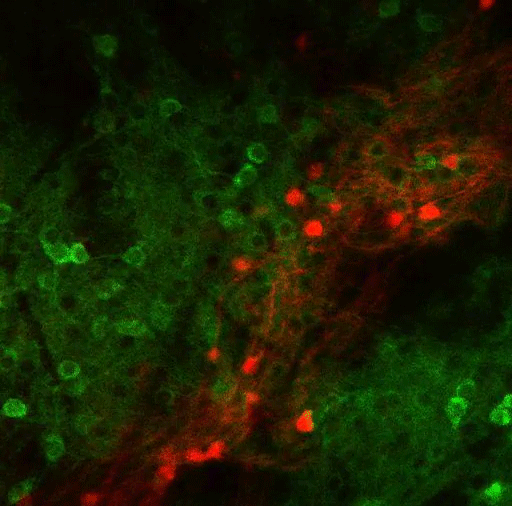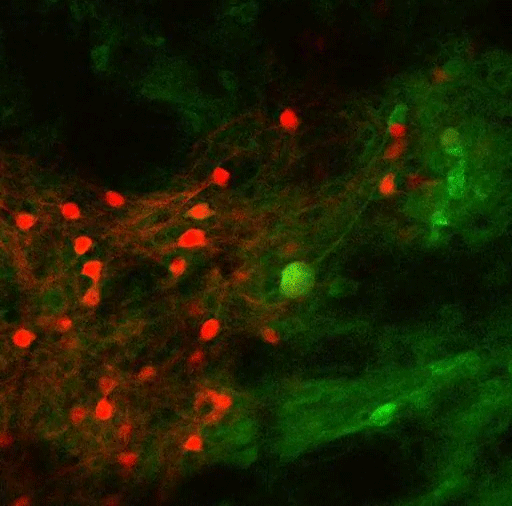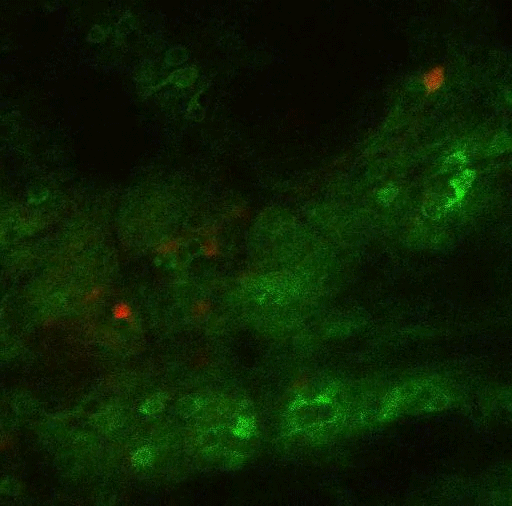
To study this kind of decision-making, the researchers trained mice to spin a wheel to the left or the right. With each turn, they would receive a combination of reward (sugary water) and negative outcome (a small puff of air). As the mice performed the task, they learned to maximize the delivery of rewards and to minimize the delivery of air puffs. However, over hundreds of trials, the researchers frequently changed the probabilities of getting the reward or the puff of air, so the mice would need to adjust their behavior.
As the mice learned to make these adjustments, the researchers recorded the activity of neurons in the striatum. They had expected to find neuronal activity that reflects which actions are good and need to be repeated, or bad and that need to be avoided. While some neurons did this, the researchers also found, to their surprise, that many neurons encoded details about the relationship between the actions and both types of outcomes.
The researchers found that these neurons responded more strongly when a behavior resulted in an unexpected outcome, that is, when turning the wheel in one direction produced the opposite outcome as it had in previous trials. These “error signals” for reward and penalty seem to help the brain figure out that it’s time to change tactics.
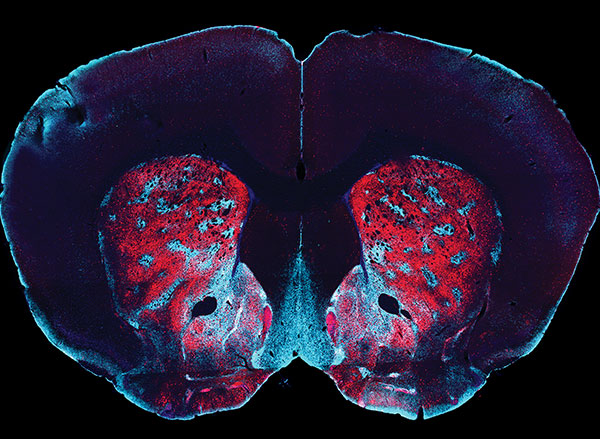
Most of the neurons that encode these error signals are found in the striosomes — clusters of neurons located in the striatum. Previous work has shown that striosomes send information to many other parts of the brain, including dopamine-producing regions and regions involved in planning movement.
“The striosomes seem to mostly keep track of what the actual outcomes are,” Bloem says. “The decision whether to do an action or not, which essentially requires integrating multiple outcomes, probably happens somewhere downstream in the brain.”
Making judgments
The findings could be relevant not only to mice learning a task, but also to many decisions that people have to make every day as they weigh the risks and benefits of each choice. Eating a big bowl of ice cream after dinner leads to immediate gratification, but it might contribute to weight gain or poor health. Deciding to have carrots instead will make you feel healthier, but you’ll miss out on the enjoyment of the sweet treat.
“From a value perspective, these can be considered equally good,” Bloem says. “What we find is that the striatum also knows why these are good, and it knows what are the benefits and the cost of each. In a way, the activity there reflects much more about the potential outcome than just how likely you are to choose it.”
This type of complex decision-making is often impaired in people with a variety of neuropsychiatric disorders, including anxiety, depression, schizophrenia, obsessive-compulsive disorder, and posttraumatic stress disorder. Drug abuse can also lead to impaired judgment and impulsivity.
“You can imagine that if things are set up this way, it wouldn’t be all that difficult to get mixed up about what is good and what is bad, because there are some neurons that fire when an outcome is good and they also fire when the outcome is bad,” Graybiel says. “Our ability to make our movements or our thoughts in what we call a normal way depends on those distinctions, and if they get blurred, it’s real trouble.”
The new findings suggest that behavioral therapy targeting the stage at which information about potential outcomes is encoded in the brain may help people who suffer from those disorders, the researchers say.
The research was funded by the National Institutes of Health/National Institute of Mental Health, the Saks Kavanaugh Foundation, the William N. and Bernice E. Bumpus Foundation, the Simons Foundation, the Nancy Lurie Marks Family Foundation, the National Eye Institute, the National Institute of Neurological Disease and Stroke, the National Science Foundation, the Simons Foundation Autism Research Initiative, and JSPS KAKENHI.