How a single gene contributes to autism and schizophrenia
McGovern neuroscientists unravel Shank3 gene's role in autism and schizophrenia.

Although it is known that psychiatric disorders have a strong genetic component, untangling the web of genes contributing to each disease is a daunting task. Scientists have found hundreds of genes that are mutated in patients with disorders such as autism, but each patient usually has only a handful of these variations.
To further complicate matters, some of these genes contribute to more than one disorder. One such gene, known as Shank3, has been linked to both autism and schizophrenia.
MIT neuroscientists have now shed some light on how a single gene can play a role in more than one disease. In a study appearing in the Dec. 10 online edition of Neuron, they revealed that two different mutations of the Shank3 gene produce some distinct molecular and behavioral effects in mice.
“This study gives a glimpse into the mechanism by which different mutations within the same gene can cause distinct defects in the brain, and may help to explain how they may contribute to different disorders,” says Guoping Feng, the James W. and Patricia Poitras Professor of Neuroscience at MIT, a member of MIT’s McGovern Institute for Brain Research, a member of the Stanley Center for Psychiatric Research at the Broad Institute, and the senior author of the study.
The findings also suggest that identifying the brain circuits affected by mutated genes linked to psychiatric disease could help scientists develop more personalized treatments for patients in the future, Feng says.
The paper’s lead authors are McGovern Institute research scientist Yang Zhou, graduate students Tobias Kaiser and Xiangyu Zhang, and research affiliate Patricia Monteiro.
Disrupted communication
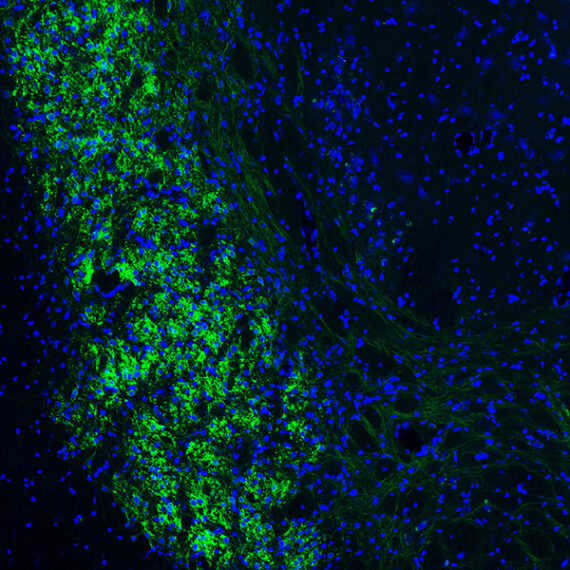
The protein encoded by Shank3 is found in synapses — the junctions between neurons that allow them to communicate with each other. Shank3 is a scaffold protein, meaning it helps to organize hundreds of other proteins clustered on the postsynaptic cell membrane, which are required to coordinate the cell’s response to signals from the presynaptic cell.
In 2011, Feng and colleagues showed that by deleting Shank3 in mice they could induce two of the most common traits of autism — avoidance of social interaction, and compulsive, repetitive behavior. A year earlier, researchers at the University of Montreal identified a Shank3 mutation in patients suffering from schizophrenia, which is characterized by hallucinations, cognitive impairment, and abnormal social behavior.
Feng wanted to find out how these two different mutations in the Shank3 gene could play a role in such different disorders. To do that, he and his colleagues engineered mice with each of the two mutations: The schizophrenia-related mutation results in a truncated version of the Shank3 protein, while the autism-linked mutation leads to a total loss of the Shank3 protein.
Behaviorally, the mice shared many defects, including strong anxiety. However, the mice with the autism mutation had very strong compulsive behavior, manifested by excessive grooming, which was rarely seen in mice with the schizophrenia mutation.
In the mice with the schizophrenia mutation, the researchers saw a type of behavior known as social dominance. These mice trimmed the whiskers and facial hair of the genetically normal mice sharing their cages, to an extreme extent. This is a typical way for mice to display their social dominance, Feng says.
By activating the mutations in different parts of the brain and at different stages of development, the researchers found that the two mutations affected brain circuits in different ways. The autism mutation exerted its effects early in development, primarily in a part of the brain known as the striatum, which is involved in coordinating motor planning, motivation, and habitual behavior. Feng believes that disruption of synapses in the striatum contributes to the compulsive behavior seen in those mice.
In mice carrying the schizophrenia-associated mutation, early development was normal, suggesting that truncated Shank3 can adequately fill in for the normal version during this stage. However, later in life, the truncated version of Shank3 interfered with synaptic functions and connections in the brain’s cortex, where executive functions such as thought and planning occur. This suggests that different segments of the protein — including the stretch that is missing in the schizophrenia-linked mutation — may be crucial for different roles, Feng says.
The new paper represents an important first step in understanding how different mutations in the same gene can lead to different diseases, says Joshua Gordon, an associate professor of psychiatry at Columbia University.
“The key is to identify how the different mutations alter brain function in different ways, as done here,” says Gordon, who was not involved in the research. “Autism strikes early in childhood, while schizophrenia typically arises in adolescence or early adulthood. The finding that the autism-associated mutation has effects at a younger age than the schizophrenia-associated mutation is particularly intriguing in this context.”
Modeling disease
Although only a small percentage of autism patients have mutations in Shank3, many other variant synaptic proteins have been associated with the disorder. Future studies should help to reveal more about the role of the many genes and mutations that contribute to autism and other disorders, Feng says. Shank3 alone has at least 40 identified mutations, he says.
“We cannot consider them all to be the same,” he says. “To really model these diseases, precisely mimicking each human mutation is critical.”
Understanding exactly how these mutations influence brain circuits should help researchers develop drugs that target those circuits and match them with the patients who would benefit most, Feng says, adding that a tremendous amount of work needs to be done to get to that point.
His lab is now investigating what happens in the earliest stages of the development of mice with the autism-related Shank3 mutation, and whether any of those effects can be reversed either during development or later in life.
The research was funded by the Simons Center for the Social Brain at MIT, the Stanley Center for Psychiatric Research at the Broad Institute of MIT and Harvard, the Poitras Center for Affective Disorders Research at MIT, and National Institute of Mental Health.