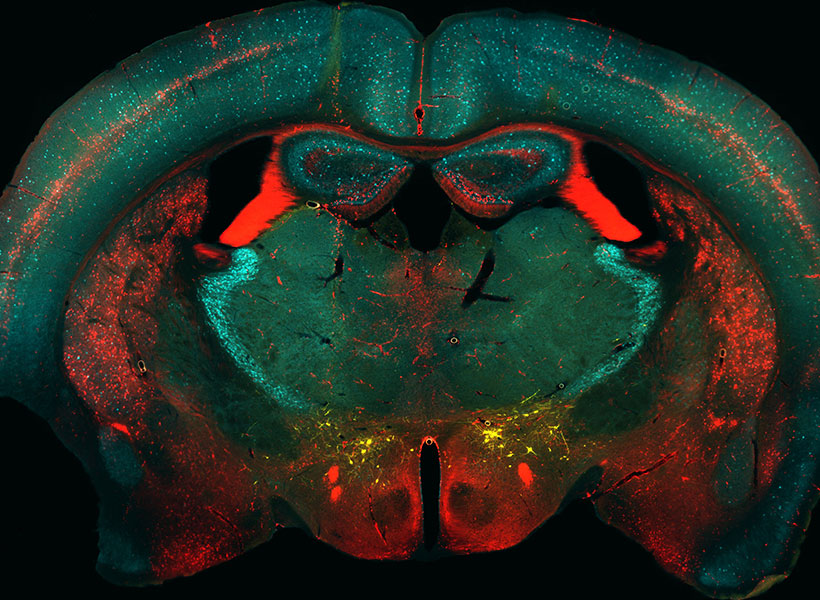
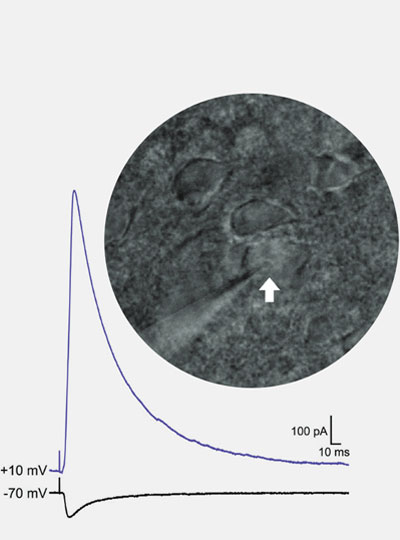
In the human brain, 86 billion neurons form more than 100 trillion connections with other neurons at junctions called synapses. Scientists at the McGovern Institute are working with their collaborators to develop technologies to map these connections across the brain, from mice to humans.
Today, the National Institutes of Health (NIH) announced a new program to support research projects that have the potential to reveal an unprecedented and dynamic picture of the connected networks in the brain. Four of these NIH-funded research projects will take place in McGovern labs.
BRAIN Initiative
In 2013, the Obama administration announced the Brain Research Through Advancing Innovative Neurotechnologies® (BRAIN) Initiative, a public-private research effort to support the development and application of new technologies to understand brain function.
Today, the NIH announced its third project supported by the BRAIN Initiative, called BRAIN Initiative Connectivity Across Scales (BRAIN CONNECTS). The new project complements two previous large-scale projects, which together aim to transform neuroscience research by generating wiring diagrams that can span entire brains across multiple species. These detailed wiring diagrams can help uncover the logic of the brain’s neural code, leading to a better understanding of how this circuitry makes us who we are and how it could be rewired to treat brain diseases.

BRAIN CONNECTS at McGovern
The initial round of BRAIN CONNECTS awards will support researchers at more than 40 university and research institutions across the globe with 11 grants totaling $150 million over five years. Four of these grants have been awarded to McGovern researchers Guoping Feng, Ila Fiete, Satra Ghosh, and Ian Wickersham, whose projects are outlined below:
BRAIN CONNECTS: Comprehensive regional projection map of marmoset with single axon and cell type resolution
Team: Guoping Feng (McGovern Institute, MIT), Partha Mitra (Cold Spring Harbor Laboratory), Xiao Wang (Broad Institute), Ian Wickersham (McGovern Institute, MIT)
Summary: This project will establish an integrated experimental-computational platform to create the first comprehensive brain-wide mesoscale connectivity map in a non-human primate (NHP), the common marmoset (Callithrix jacchus). It will do so by tracing axonal projections of RNA barcode-identified neurons brain-wide in the marmoset, utilizing a sequencing-based imaging method that also permits simultaneous transcriptomic cell typing of the identified neurons. This work will help bridge the gap between brain-wide mesoscale connectivity data available for the mouse from a decade of mapping efforts using modern techniques and the absence of comparable data in humans and NHPs.
BRAIN CONNECTS: A center for high-throughput integrative mouse connectomics
Team: Jeff Lichtman (Harvard University), Ila Fiete (McGovern Institute, MIT), Sebastian Seung (Princeton University), David Tank (Princeton University), Hongkui Zeng (Allen Institute), Viren Jain (Google), Greg Jeffries (Oxford University)
Summary: This project aims to produce a large-scale synapse-level brain map (connectome) that includes all the main areas of the mouse hippocampus. This region is of clinical interest because it is an essential part of the circuit underlying spatial navigation and memory and the earliest impairments and degeneration related to Alzheimer’s disease.
BRAIN CONNECTS: The center for Large-scale Imaging of Neural Circuits (LINC)
Team: Anastasia Yendiki (MGH), Satra Ghosh (McGovern, MIT), Suzanne Haber (University of Rochester), Elizabeth Hillman (Columbia University)
Summary: This project will generate connectional diagrams of the monkey and human brain at unprecedented resolutions. These diagrams will be linked both to the neuroanatomic literature and to in vivo neuroimaging techniques, bridging between the rigor of the former and the clinical relevance of the latter. The data to be generated by this project will advance our understanding of brain circuits that are implicated in motor and psychiatric disorders, and that are targeted by deep-brain stimulation to treat these disorders.
BRAIN CONNECTS: Mapping brain-wide connectivity of neuronal types using barcoded connectomics
Team: Xiaoyin Chen (Allen Institute), Ian Wickersham (McGovern Institute, MIT), and Justus Kebschull of JHU
Summary: This project aims to optimize and develop barcode sequencing-based neuroanatomical techniques to achieve brain-wide, high-throughput, highly multiplexed mapping of axonal projections and synaptic connectivity of neuronal types at cellular resolution in primate brains. The team will work together to apply these techniques to generate an unprecedented multi-resolution map of brain-wide projections and synaptic inputs of neurons in the macaque visual cortex at cellular resolution.